Transposonmapper output data postprocessing
Contents
Transposonmapper output data postprocessing¶
## Importing the required python libraries
import os, sys
import warnings
import timeit
import numpy as np
import pandas as pd
import pkg_resources
How to clean the wig and bed files¶
Here we will remove transposon insertions in .bed and .wig files that were mapped outside the chromosomes, creates consistent naming for chromosomes and change the header of files with custom headers.
Clean wig files for proper visualization in the genome Browser http://genome-euro.ucsc.edu/cgi-bin/hgGateway
from transposonmapper.processing.clean_bedwigfiles import cleanfiles
######## Lets save the wig and bed files as variables to clean them and call the function#####################
import glob
wig_files=[]
bed_files=[]
data_dir = pkg_resources.resource_filename("transposonmapper", "data_files/files4test/")
#data_dir="../transposonmapper/data_files/files4test/"
wig_files = glob.glob(os.path.join(data_dir, '*sorted.bam.wig'))
bed_files = glob.glob(os.path.join(data_dir, '*sorted.bam.bed'))
############## Cleaning the files #############################
custom_header = ""
split_chromosomes = False
for files in zip(wig_files,bed_files):
cleanfiles(filepath=files[0], custom_header=custom_header, split_chromosomes=split_chromosomes)
cleanfiles(filepath=files[1], custom_header=custom_header, split_chromosomes=split_chromosomes)
Wig file loaded /data/localhome/linigodelacruz/Documents/PhD_2018/Documentation/SATAY/src(source-code)/Transposonmapper/transposonmapper/data_files/files4test/SRR062634.filt_trimmed.sorted.bam.wig
evaluating chromosome I
evaluating chromosome II
evaluating chromosome III
evaluating chromosome IV
evaluating chromosome V
evaluating chromosome VI
evaluating chromosome VII
evaluating chromosome VIII
evaluating chromosome IX
evaluating chromosome X
evaluating chromosome XI
evaluating chromosome XII
evaluating chromosome XIII
evaluating chromosome XIV
evaluating chromosome XV
evaluating chromosome XVI
Bed file loaded /data/localhome/linigodelacruz/Documents/PhD_2018/Documentation/SATAY/src(source-code)/Transposonmapper/transposonmapper/data_files/files4test/SRR062634.filt_trimmed.sorted.bam.bed
evaluating chromosome I
evaluating chromosome II
evaluating chromosome III
evaluating chromosome IV
evaluating chromosome V
evaluating chromosome VI
evaluating chromosome VII
evaluating chromosome VIII
evaluating chromosome IX
evaluating chromosome X
evaluating chromosome XI
evaluating chromosome XII
evaluating chromosome XIII
evaluating chromosome XIV
evaluating chromosome XV
evaluating chromosome XVI
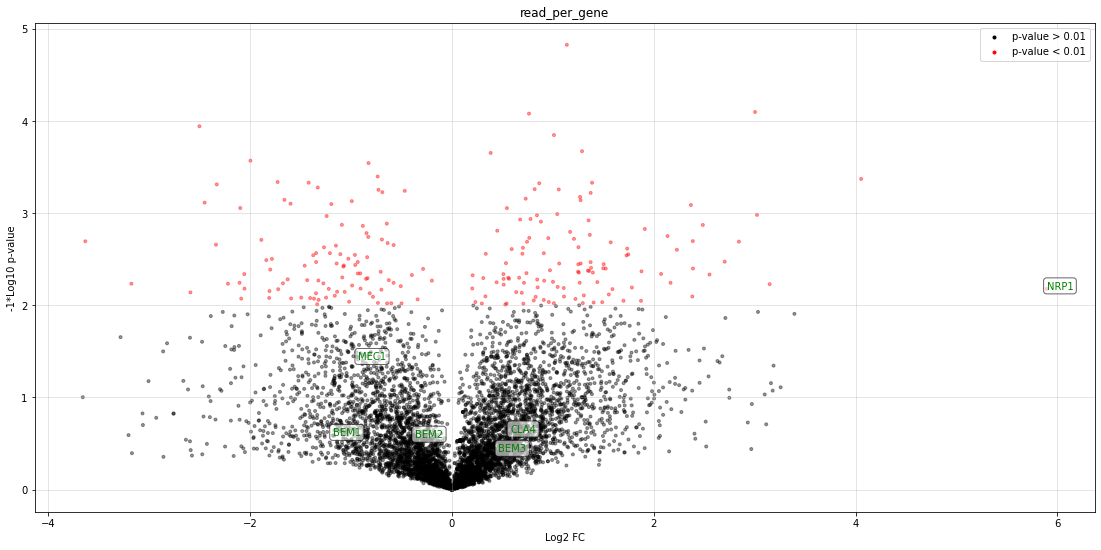
Visualize the insertions and reads per gene throughout the genome¶
## Import the function
from transposonmapper.processing.transposonread_profileplot_genome import profile_genome
####Lets save the cleaned files as variables to clean them and call the function####
cleanbed_files=[]
for root, dirs, files in os.walk(data_dir):
for file in files:
if file.endswith("clean.bed"):
cleanbed_files.append(os.path.join(root, file))
cleanwig_files=[]
for root, dirs, files in os.walk(data_dir):
for file in files:
if file.endswith("clean.wig"):
cleanwig_files.append(os.path.join(root, file))
#### vizualization #####
bed_file=cleanbed_files[0] # example for the 1st file
variable="transposons" #"reads" "transposons"
bar_width=None
savefig=False
profile=profile_genome(bed_file=bed_file, variable=variable, bar_width=bar_width, savefig=savefig,showfig=True)
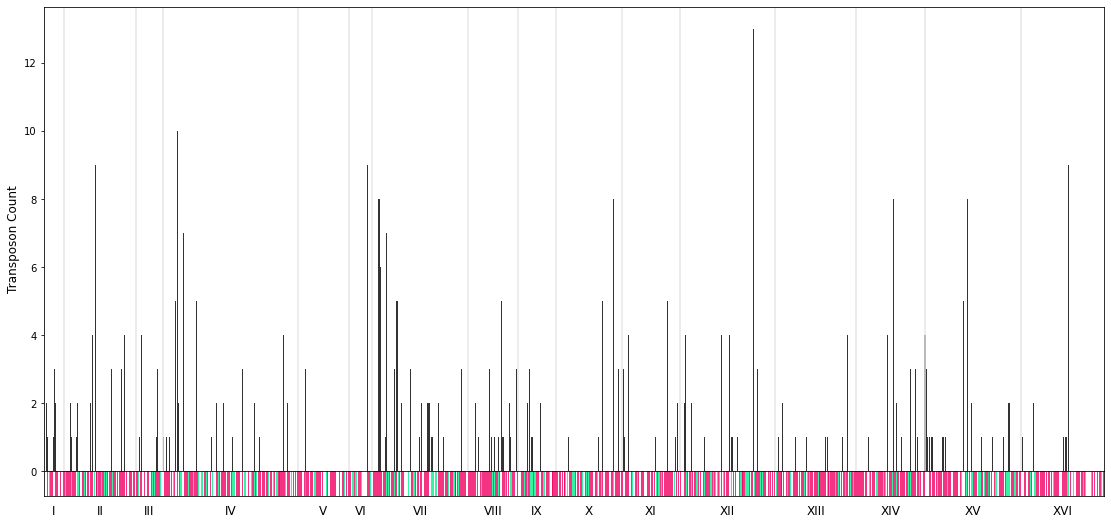
Zoom in into the chromosomes¶
from transposonmapper.processing.genomicfeatures_dataframe import dna_features
##### getting the files #########
pergene_files=[]
data_dir = pkg_resources.resource_filename("transposonmapper", "data_files/files4test/")
# data_dir="../transposonmapper/data_files/files4test/"
for root, dirs, files in os.walk(data_dir):
for file in files:
if file.endswith('sorted.bam_pergene_insertions.txt'):
pergene_files.append(os.path.join(root, file))
#### vizualization #####
wig_file = cleanwig_files[0]
pergene_insertions_file = pergene_files[0]
plotting=True
variable="reads" #"reads" or "insertions"
savefigure=False
verbose=True
region = "I" #e.g. 1, "I", ["I", 0, 10000"], gene name (e.g. "CDC42")
dna_features(region=region,
wig_file=wig_file,
pergene_insertions_file=pergene_insertions_file,
variable=variable,
plotting=plotting,
savefigure=savefigure,
verbose=verbose)
This is the plot for the case of the dummy sample files for chromosome I.
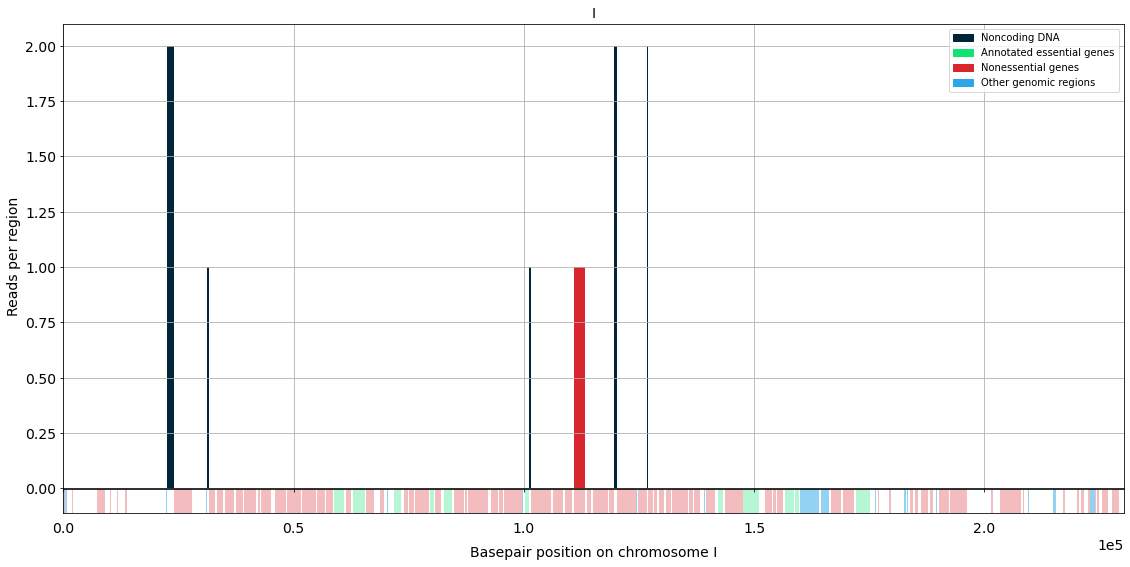
Volcano plots¶
Do you want to compare two differente libraries to discover which genes stood out from their comparison?
Then do volcano plots!!
Getting the volcano plot¶
Look at the help of this function , HERE
from transposonmapper.statistics import volcano
# Please be aware that you should add the location of your tab separated pergene files (output of the pipeline)
# to the volcano function from
# two libraries.
# And also be aware you will need at least two replicates per library in order to have statistics
# for the volcano plot.
path_a = r""
filelist_a = ["",""]
path_b = r""
filelist_b = ["",""]
variable = 'read_per_gene' #'read_per_gene' 'tn_per_gene', 'Nreadsperinsrt'
significance_threshold = 0.01 #set threshold above which p-values are regarded significant
normalize=True
trackgene_list = ['my-favorite-gene'] # ["cdc42"]
figure_title = " "
volcano_df = volcano(path_a=path_a, filelist_a=filelist_a,
path_b=path_b, filelist_b=filelist_b,
variable=variable,
significance_threshold=significance_threshold,
normalize=normalize,
trackgene_list=trackgene_list,
figure_title=figure_title)
This is a volcano plot made with real data!¶
Comparing the libraries of wild type vs \(\Delta\) nrp1