Tutorial-4: Zoom in into the chromosomes¶
Here we will use the files generated in Tutorial-2 : How to clean the wig and bed files..
Import the function¶
from transposonmapper.processing.genomicfeatures_dataframe import dna_features
Getting the pergene file¶
pergene_files=[]
data_dir="../transposonmapper/data_files/files4test/"
for root, dirs, files in os.walk(data_dir):
for file in files:
if file.endswith('sorted.bam_pergene_insertions.txt'):
pergene_files.append(os.path.join(root, file))
Vizualization¶
wig_file = cleanwig_files[0]
pergene_insertions_file = pergene_files[0]
plotting=True
variable="reads" #"reads" or "insertions"
savefigure=False
verbose=True
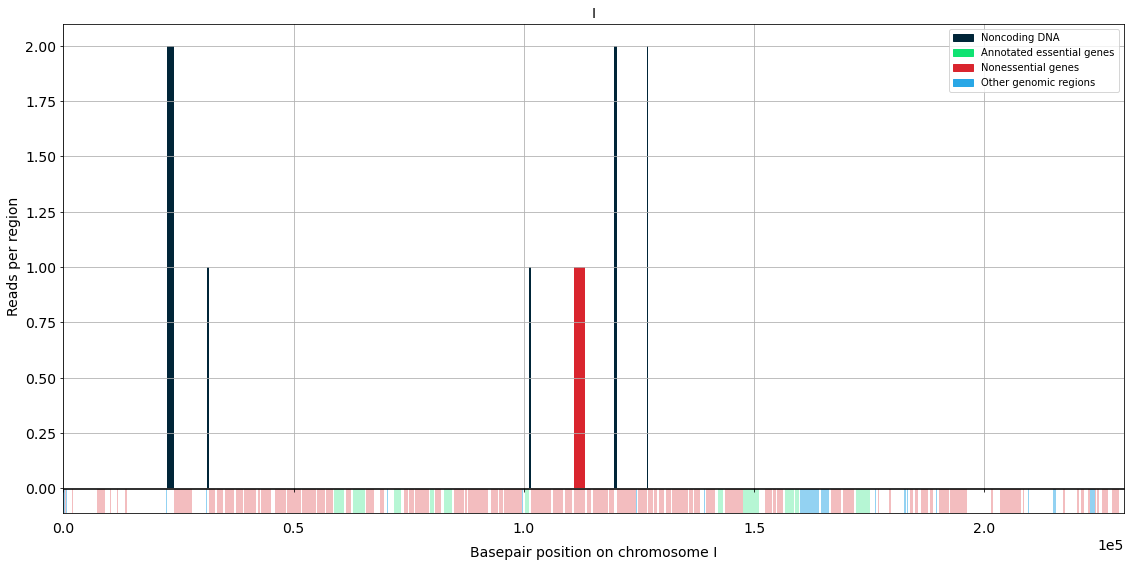
region = "I" #e.g. 1, "I", ["I", 0, 10000"], gene name (e.g. "CDC42")
dna_features(region=region,
wig_file=wig_file,
pergene_insertions_file=pergene_insertions_file,
variable=variable,
plotting=plotting,
savefigure=savefigure,
verbose=verbose)
This will create a dataframe with the following columns per region:
Feature_name
Standard_name
Feature_alias
Feature_type
Essentiality
Chromosome
Position
Nbasepairs
Ninsertions
Ninsertions_truncatedgene
Nreads
Nreads_list
Nreads_truncatedgene
Nreadsperinsrt
Nreadsperinsrt_truncatedgene
This is the plot for the case of the dummy sample files for chromosome I.